Allele-specific expression (ASE) analysis, which quantifies the relative expression of two alleles in a diploid individual, is a powerful tool for identifying cis-regulated gene expression variations that underlie phenotypic differences among individuals. To understand the role of ASE in human diseases, we exploit allelic imbalance by RNA-seq, which provides allele-specific read counts at exonic SNPs distinguished by heterozygous sites. Evidence of ASE is often shared across multiple SNPs within the same gene and across multiple individuals. We are interested in developing methods to model gene-level ASE across SNPs and individuals, and to detect gene-level ASE under one condition and differential ASE between two conditions (e.g., pre- vs post-treatment).
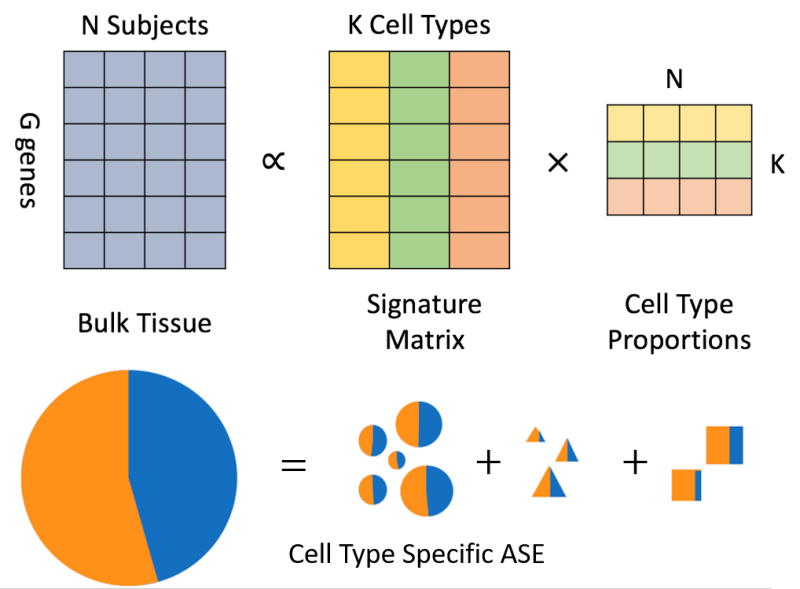
ASE detection is often performed in bulk RNA-seq data, a data type that averages out possible heterogeneity in a mixture of different cell types. Since ASE may vary across different cell types, with the recent advance in single-cell RNA sequencing (scRNA-seq) technologies, characterizing ASE at the cell type resolution may help reveal more about the gene regulation. However, scRNA-seq data is costly to generate and noisy with excessive zeros due to transcriptional bursting. Therefore, it is desirable to incorporate information obtained from single-cell data to characterize cell-type specific ASE. We are interested in developing statistical methods to infer cell-type specific ASE, by integrating scRNA-seq data and bulk RNA-seq data.